![]()

![]()
Al pulsar esta opción damos paso al visualizador de moléculas. Aparece una ventana donde muestra al usuario el sistema de ficheros y permite seleccionar el fichero que contiene la molécula que queremos visualizar. Permite ficheros que tengan la extensión [*.mol], [*.mld] y [*.xyz], para más información respecto el formato de estos ficheros ver Formatos aceptados.
Al pulsar esta opción damos paso al visualizador de moléculas. Con esta opción se puede ver un conjunto de moléculas ya predefinidas para que las pueda seleccionar el usuario y permita ya un uso del visualizador.
Con esta opción damos paso al editor de cristales, en este caso seguimos los esquemas de las Redes de Bravais. Dado que hay siete sistemas (Monoclínico. Triclínico, Ortorrómbico, Trigonal, Tetragonal, Hexagonal, Cúbico) con sus respectivas variedades, hay un menú desplegable por cada sistema. A partir de este momento se activan los botones/opciones de "Añadir átomo", "Añadir enlace", "seleccionar nuevo átomo", "Cristalizar" del editor. La opción que da por finalizada la creación del cristal es la opción cristalizar, donde lo pasamos al visualizador de moléculas, que en este caso visualizará un cristal, y las opciones de creación de cristales se desactivarán.
Con esta opción damos paso al editor de cristales. En esta ocasión pasamos a estudiar los cristales conocidos como Cristales Simples. Tal como pasaba con la anterior opción, se activan los botones/opciones que permiten la creación de cristales, "Añadir átomo", "Añadir enlace", "seleccionar nuevo átomo", "Cristalizar" del editor. La opción que da por finalizada la creación del cristal es la opción cristalizar, donde lo pasamos al visualizador de moléculas, que en este caso visualizará un cristal, y las opciones de creación de cristales se desactivarán.
Con esta opción damos paso al visor de planos cristalográficos. Los planos cristalográficos sólo tienen sentido si los aplicamos a los cristales, por esa razón sólo estará activo si lo que tenemos en el visualizador es un cristal.
Los parámetros (h, k, l) representan los índices de Miller, o sea, el inverso de los puntos de intersección del plano con los ejes de coordenadas (x,y,z) respectivamente.
Con esta opción grabaremos el fichero en el disco duro. Estará activa si hay alguna molécula activa en la aplicación. Hay que tener en cuenta que solo podrá grabar la molécula si antes de la ejecución de la aplicación le hemos dado permisos de escritura al disco duro. Para más información ver Permisos.
Con esta opción grabaremos el fichero en el disco duro. Surgirá una ventana donde indicaremos el directorio y el fichero sobre el que deseamos grabar la molécula. Es obligatorio poner la extensión del formato que deseamos grabar. Hay tres formatos: [*.mol] [*.mld] [*.xyz]. Para más información sobre los formatos ver Formatos aceptados. Hay que tener en cuenta que solo podrá grabar la molécula si antes de la ejecución de la aplicación le hemos dado permisos de escritura al disco duro. Para más información ver Permisos.
Con esta opción imprimiremos la molécula que se vea por pantalla. Es necesario para imprimir que antes el usuario haya dado los privilegios de impresión a la aplicación. Para más información ver Permisos.
Con esta opción daremos por acabada la sesión con la aplicación, y saldremos de ella.
![]()
Las siguientes opciones sólo se activan en el caso de que estemos en la edición de cristales.
Al seleccionar está opción estará activo el estado de añadir átomos. Para añadir un átomo nos hemos de situar con el cursor encima de un vértice del modelo que representa la malla de un posible cristal y realizar un único clic con el ratón. Si queremos deseleccionar el átomo/vértice volvemos a hacer un clic encima del átomo que queremos quitar.
Al seleccionar esta opción estará activo el estado de añadir enlaces. Para añadir un enlace nos hemos de situar con el cursor encima de la arista que deseamos convertir en enlace y hacer un clic con el ratón. Si queremos quitar el enlace nos pondremos encima del enlace que queremos quitar y haremos un clic.
Al seleccionar esta opción aparecerá la siguiente ventana donde se podrá seleccionar que tipo de átomo queremos insertar a partir de este momento. Por defecto siempre estará primero activo el Carbono.
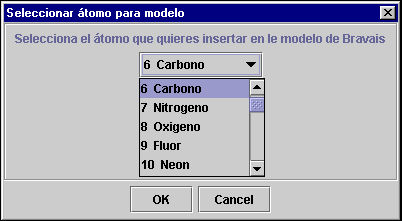
Cuando pulsemos esta opción estamos indicando que el modelo que hemos hecho ya esta listo para ser cristalizado, y así poder ser visto en el visualizador de moléculas. Dado que hemos hecho la celda unitaria del cristal, primero hemos de replicar este cristal tantas veces como queramos, en la ventana que sigue a continuación hemos de poner el numero de celdas adyacentes a lo largo, ancho y alto, o en los ejes Z, X, Y, respectivamente. Los números que se inserte en los tres campos tendrán que ser siempre números naturales superiores a cero.

![]()
Con la opción zoom lo que se hace es ampliar la imagen respecto a la imagen anterior. Para hacer zoom nos situamos con el cursor sobre la zona de la imagen que deseamos ver con más detalle y hacemos un clic, la imagen que aparecerá nueva estará ampliada un 20% más que la anterior y centrada en el punto donde hicimos clic. Sin embargo existe un límite a partir del cual realizar más zoom degenera la imagen, no se puede hacer zoom para escenas más pequeñas de 0.009 Å
Unzoom es la operación inversa de la operación Zoom. Cada vez que el usuario dé un clic sobre la molécula, empequeñecerá un 20% respecto la anterior.
La operación Arrastrar nos permite mover la molécula por el área de visualización. Para mover la figura hemos de pulsar el ratón y mantenerlo pulsado mientras lo movemos por la pantalla.
Cuando está opción está activa permite girar la molécula. Para girar la molécula se ha de apretar el botón del ratón encima de la molécula y mantenerlo pulsado mientras al mismo tiempo movemos el ratón.
Si por alguna motivo la escena que se visualiza degenera, pulsando esta opción vuelve a situar la escena correctamente dibujada y centrada, pudiéndose ver toda la escena en la pantalla.
Existen tres direcciones privilegiadas para ver la molécula, que corresponden con los ejes X, Y, Z, o lo que es lo mismo, ver la escena vista desde el alzado, planta y perfil, respectivamente. En esta caso al elegir esta opción veremos la escena desde el alzado.
En esta caso al elegir esta opción veremos la escena desde la planta, vista la molécula desde ele eje Y.
En esta caso al elegir esta opción veremos la escena desde el perfil, o desde el eje Z.
![]()
Con esta opción vamos a configurar la aplicación según los parámetros que deseemos. Cuando pulsemos este botón surgirá una ventana con cuatro solapas. A continuación nos detendremos en cada solapa:
Solapa Visualización:
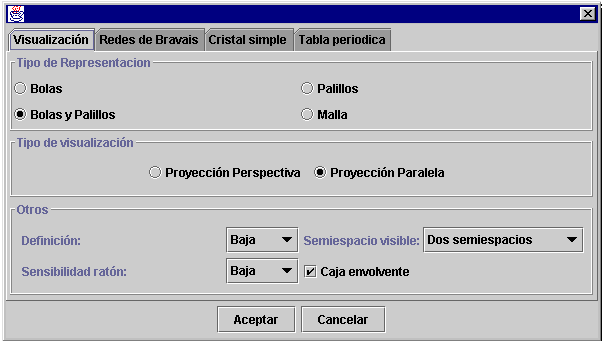
Esta solapa está dedicada a los parámetros de visualización. En la aplicación existen tres vistas, el visualizador de moléculas, el editor de cristales y el visor de Planos cristalográficos, por lo tanto cada vista tiene sus propios parámetros de visualización, cuando aparece la solapa visualización corresponderá a la vista que esté en uso en ese momento:
![]() Campo
Tipo representación: En esta campo existen cuatro tipos posibles:
Campo
Tipo representación: En esta campo existen cuatro tipos posibles:
![]() Campo
Tipo visualización:
Campo
Tipo visualización:
![]() Campo
Otros:
Campo
Otros:
Solapa Redes de Bravais:

En esta solapa pondremos los valores por defecto de las Redes de Bravais. Los valores de A, B, C representan el tamaño de las aristas del paralelepípedo. Los valores que se introducen han de estar en angstroms. Los ángulos Alfa, Beta, Gamma, es el ángulo entre las aristas del paralelepípedo, B-C, A-C, A-B, respectivamente, y el valor de los ángulos han de estar en grados.
Solapa Cristal simpl:e
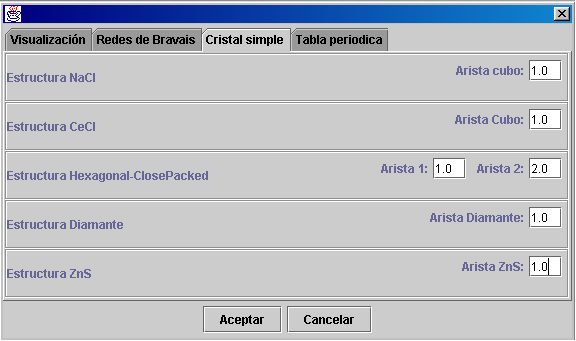
En esta solapa pondremos los valores por defecto de las estructuras de cristal simple. Los valores que se introducirán han de estar en angstroms.
Solapa Tabla periódica:
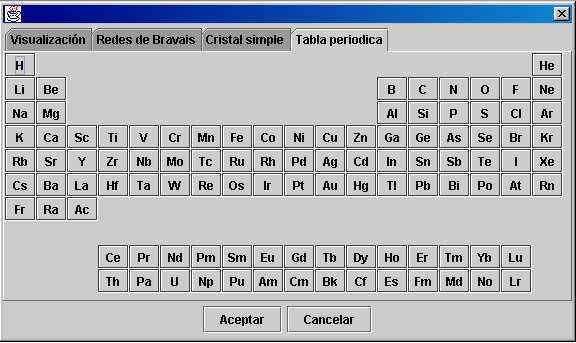
En esta solapa aparece la tabla periódica. Cada vez que deseemos cambiar el color o el radio covalente de un elemento, pulsaremos con un clic en el elemento y aparecerá la siguiente ventana:

Donde surgirá la información del átomo. Si queremos cambiar el color pulsamos el botón que tiene como etiqueta Color, y saldrá una ventana donde podremos escoger el color que deseemos. Y si lo que queremos es cambiar el radio covalente, pondremos el número que deseemos, este numero tendrá que estar en angstroms. La información que pongamos será la misma para todas las moléculas.
![]()
La opción acerca de muestra la siguiente ventana
